Die DNA ist mit einer Länge von bis zu 10 cm (menschliches Chromosom 1) das größte Biopolymer der Natur. Sie ist in einer komplexen und hoch organisierten Struktur - dem Chromatin - gebündelt, um eine effiziente Verpackung im Zellkern zu gewährleisten, was erhebliche Auswirkungen auf die Genfunktion und -regulation hat. Unser Verständnis der DNA-Verpackung und der Regulationsmechanismen, die über große Entfernungen (bis zu mehreren Megabasenpaaren) wirksam werden, ist jedoch begrenzt. Einer der Gründe dafür ist, dass die Modellierung von DNA aufgrund ihrer Größe und Komplexität besonders schwierig ist. In der Biophysik liefern Berechnungswerkzeuge wie die Molekulardynamik strukturelle Erkenntnisse nur auf der kleinräumigen atomistischen Ebene. In der Hochdurchsatz-Genomik bieten die Methoden zur Erfassung der Chromosomenkonformation lediglich grobkörnige Strukturinformationen, die sich auf paarweise Kontaktwahrscheinlichkeiten beschränken. Um unser Verständnis der 3D-Struktur der DNA zu vertiefen, planen wir, Ergebnisse aus Bottom-up- und Top-down-Ansätzen zu integrieren. Wir werden große Mengen an Sequenzierungsdaten generieren, um Chromatin-Interaktionsmatrizen in verschiedenen Zelltypen während des Entwicklungsstadiums sowie von Patienten mit Chromosomenaberrationen zu erstellen, die bekanntermaßen neurologische Entwicklungsstörungen verursachen. Dabei werden wir auch den Geschlechtsdimorphismus berücksichtigen. Experimentell wollen wir die Stärken der Long-Read-Sequenzierung für die Erfassung der Chromatinkonformation nutzen, um 3D-Strukturen, komplexe Umlagerungen und strukturelle Varianten aufzulösen. Anschließend werden wir diese experimentellen Daten nutzen, um Chromatinmodelle mit Vorhersagekraft zu konstruieren, die auf Konzepten aus molekularen Simulationen grobkörniger Polymere basieren. Parallel dazu werden wir durch gezielte Simulationen zusätzliche Parameter (z. B. epigenetische Faktoren) ermitteln, die die Chromatinfaltung beeinflussen. In einer 12-Jahres-Perspektive wollen wir verstehen, wie mikroskopische Modifikationen (z. B. kleine Mutationen, genomische Varianten und epigenetische Veränderungen, die sich von Zelle zu Zelle unterscheiden) signifikante globale makroskopische Strukturveränderungen hervorrufen und wie diese zu menschlichen Krankheiten führen können. Zu diesem Zweck wollen wir die Sequenzierung und Modellierung bis hinunter auf die Einzelzellebene etablieren.
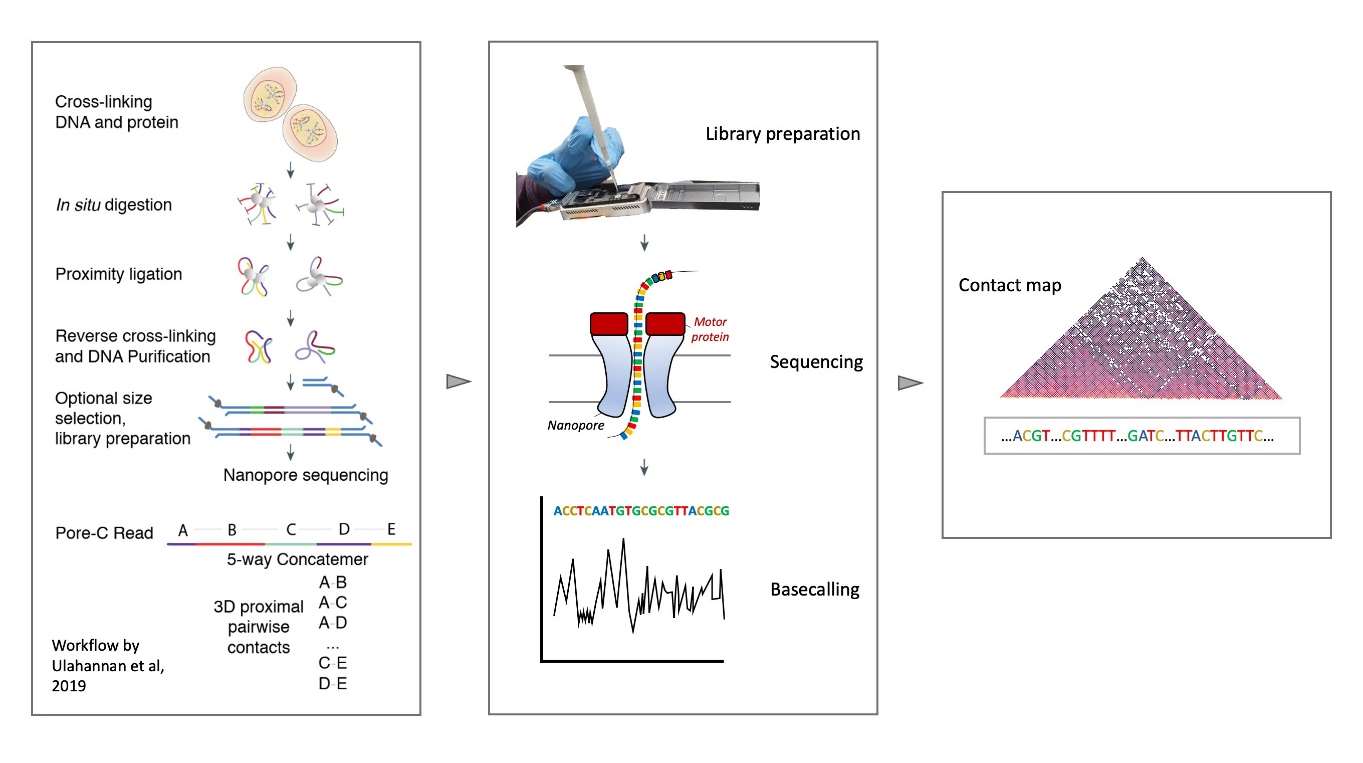
Schematische Darstellung der Pore-C-Technologie. Das Protokoll ist dem Hi-C-Protokoll ähnlich: DNA und Histone werden so vernetzt, dass die räumliche Nähe der interagierenden Loci erhalten bleibt. Durch Restriktionsverdau und anschließende Proximity-Ligation werden vernetzte Fragmente aus mehreren interagierenden Loci zusammengefügt. Diese Fragmente werden mit der ONT-Technologie sequenziert. Durch Zuordnung jedes einzelnen Alignments zu einem einzelnen Restriktionsfragment wird eine Mehrweg-Kontaktmatrix erstellt, die in paarweise Kontakte zerlegt werden kann.