Ein weiteres Papier des SFB1551 ist erschienen! "Sequenzdeterminanten der Phasentrennung von Proteinen und Erkennung durch phasengetrennte Proteinkondensate durch molekulare Dynamik und aktives Lernen" wurde auf Faraday Discussions veröffentlicht.
Abstrakt
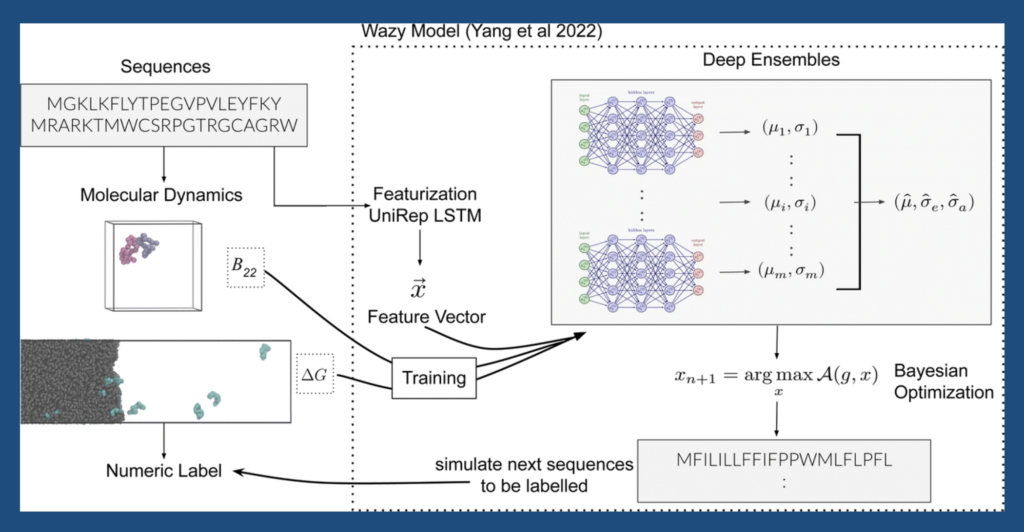
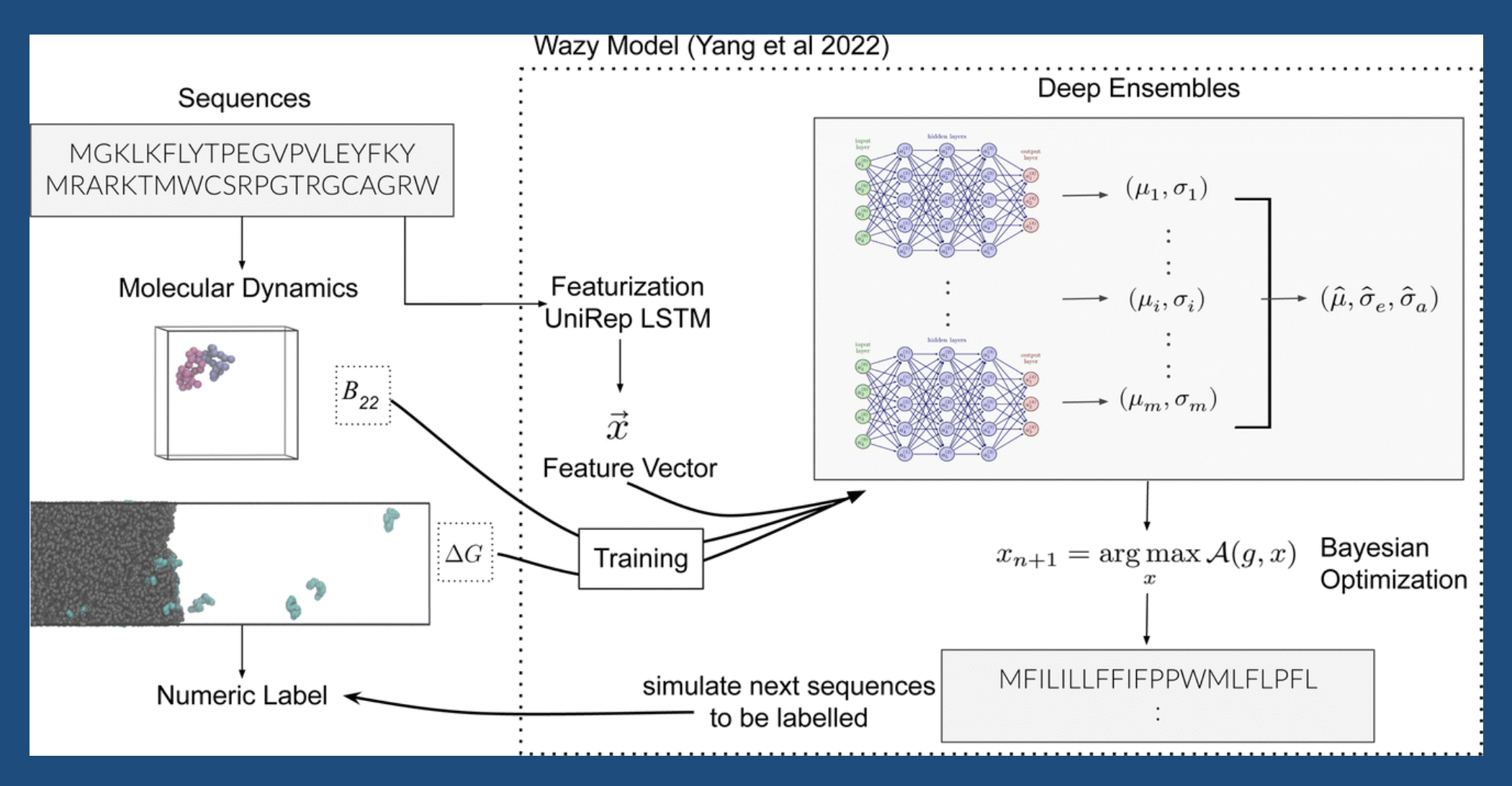
"Die Klärung der Frage, wie die Proteinsequenz die Eigenschaften ungeordneter Proteine und ihrer phasengetrennten Kondensate bestimmt, ist eine große Herausforderung für die computergestützte Chemie, Biologie und Biophysik. Quantitative Molekulardynamiksimulationen und abgeleitete freie Energiewerte können im Prinzip erfassen, wie eine Sequenz die chemischen und biologischen Eigenschaften eines Proteins kodiert. Diese Berechnungen sind jedoch sehr rechenintensiv, selbst wenn man die Darstellung durch Grobkörnigkeit reduziert; die Erkundung der großen Räume potenziell relevanter Sequenzen bleibt eine gewaltige Aufgabe. Wir verwenden ein "aktives Lernverfahren", das von Yang et al. (bioRxiv, 2022, https://doi.org/10.1101/2022.08.05.502972), um die Anzahl der benötigten markierten Beispiele aus Simulationen zu reduzieren, wobei ein auf einem neuronalen Netz basierendes Modell die nützlichsten Beispiele für den nächsten Trainingszyklus vorschlägt. Unter Anwendung dieses Bayes'schen Optimierungsrahmens bestimmen wir Eigenschaften von Proteinsequenzen mit grobkörniger Molekulardynamik, was es dem Netzwerk ermöglicht, Sequenz-Eigenschafts-Beziehungen für ungeordnete Proteine und ihre Selbstwechselwirkungen sowie ihre Wechselwirkungen in phasengetrennten Kondensaten herzustellen. Wir zeigen, wie das iterative Training mit zweiten Virialkoeffizienten, die aus den Simulationen ungeordneter Proteinsequenzen abgeleitet wurden, zu einer schnellen Verbesserung der Vorhersage von Peptid-Selbstwechselwirkungen führt. Wir verwenden diesen Bayes'schen Ansatz, um effizient nach neuen Sequenzen zu suchen, die an Kondensate der ungeordneten C-terminalen Domäne (CTD) der RNA-Polymerase II binden, indem wir die molekulare Erkennung von Peptiden an phasengetrennte Kondensate in grobkörniger Molekulardynamik simulieren. Durch die Suche nach Proteinsequenzen, die lieber mit sich selbst als mit einer anderen Proteinsequenz interagieren, können wir die Morphologie von Proteinkondensaten gestalten und mehrphasige Proteinkondensate entwerfen."
Herzlichen Glückwunsch an Lukas Stelzl (JGU, IMB) & Jan Padeken (IMB) Labs zu ihrer Veröffentlichung. Gut gemacht!